Description CovReport2 is a tool to visualize the coverage data of a diagnostic sequencing test.
It generates a one-glance overview of exon-level coverage for a given BAM or CRAM file and a list of genes (or/and transcripts) of interest.
The resulting document in a PDF format can be directly annexed to a diagnostic report.
The article describing the first release of this tool was published in
Scientific Reports A new tool CovReport generates easy-to-understand sequencing coverage summary for diagnostic reports Sci Rep 10-Apr-2020; 10(1):6247; DOI: 10.1038/s41598-020-63079-4; PMID: 32277129
The input for the first release of CovReport was a coverage file generated by an external tool.
The second release is a major upgrade as it now uses BAM or CRAM files as a direct input.
This significantly simplifies data preparation and processing by removing dependency on third party tools.
The CovReport2 has been implemented as a standalone Java application and can run on any platform where
Java Runtime (JRE) is installed (Windows, Mac or Linux). The supported Java version is 8.
Background
There is a critical need to make diagnostic reports of high-throughput sequencing extremely clear
to avoid any misinterpretation. Target sequence coverage is a key quality control information for
a sequencing test, since important clinical decisions are made based on this data. If coverage
is not sufficient even for a small region of a highly suspected candidate gene, an additional
sequencing test is needed to make sure that the pathogenic variant is not missed.
Design
The application creates PDF reports by performing the following steps:
Step 1. Create a genomic interval dataset from the user specified list of genes or/and transcripts.
There is an option to save this dataset as a BED file
(see details in Work files section).
This step uses the RefSeqExon file obtained from UCSC Genome Browser
(see details in Assembly mapping section).
The dataset is generated for the genome version hg19 or hg38 based on the assembly property in the input BAM/CRAM file.
Step 2. Create a read depth histogram for each genomic region entry in the genomic interval dataset.
This step scans records in the input BAM/CRAM file and updates the dataset with a histogram.
Depth for each genomic position is calculated based on CIGAR string information.
Only M(Match or mismatch), EQ(Matches the reference) or X(Mismatches the reference) are counted towards coverage.
Step 3. Create coverage data.
This step uses the histogram data to generate the percent covered value for each exon based on the user specified depth
(see details in Configuration section).
Step 4. Create PDF report.
The process time depends on BAM/CRAM file size and the number of genes/transcripts specified for the report.
The process status is displayed in a popup progress bar with option to cancel the process at any time.
Adjustments to the PDF report format can be made without lengthy BAM/CRAM file re-processing.
Downloadcounter Click here
to download CovReport2 application in a compressed format.
Make sure that you agree with Terms and Condition before you start to download or/and use the Software.
We are currently working on making CovReport2 even faster.
If you would like to be notified when this version is updated please send us an email to info@jdotsoft.com.
Provide your contact information to receive email about new release.
The downloaded compressed file CovReport2.zip should be extracted into local hard drive deployment folder, for example, C:\CovReport2.
The content of this folder will be the following:
msg - folder with internalization message files used for generated PDF file
(see also Language section)
RefSeqExons - folder with standard hg19 and hg38 assembly files
CovReport2.jar - Java executable archive
run.cmd - Windows command to start the application
runCommandLine.cmd - Windows command line helper
After the first execution of CovReport2 application the following items will be created in the deployment folder:
pdf-results - folder with PDF files generated by the application
CovReport2.config - file with configuration data;
this file could be manually updated, moved and reused to replace default one for command line execution
User Interface (UI)
After launching the application the following minimum steps are required to generate a PDF report:
Load a BAM or CRAM file
Load a file with a list of genes or/and transcripts (plain text format, one line per gene or transcript)
Enter patient name
Generate report
The example of generated PDF file could be found here.
Command line
The application could also be started via command line with the following options:
-h,--help help
-i,--input <arg> input BAM or CRAM file
-g,--genes <arg> input Gene&Transcript list file
-p,--patient <arg> patient name
-r <arg> RefSeqExon file (optional)
-comments <arg> comments (optional)
-d,--dir <arg> directory for generated PDF file (optional)
-f,--filename <arg> generated PDF filename (optional)
-config <arg> config file (optional)
The optional config file parameter is CovReport2.config generated by the application started in UI mode.
It could be edited manually and moved to any folder.
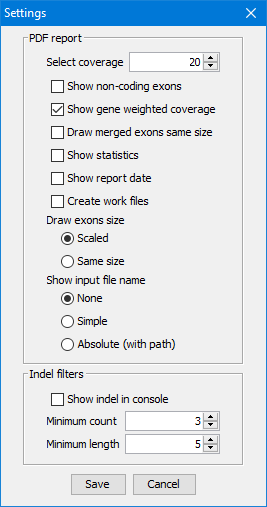
Configuration
The application could be configured with the following two sets of options:
Main window - options which are modified often (see User Interface (UI) section):
Skip white genes
Merge white exons
Merge non-white exons
Show genes transcripts
Settings window - options which are modified rarely (see Configuration section):
Choose coverage
Show non-coding exons
Show gene weighted coverage
Draw merged exons same size
Show statistics
Show report date
Create work files
Draw exon size as scaled or all exons the same size
Show input file name { none, simple, absolute file name }
All configuration options are saved in CovReport2.config file for the next execution.
Click the button in the toolbar to open Settings popup
(screenshot below for the default values):
Language
All text in the generated PDF file is in the current locale language.
The following languages are supported in the current distribution: English and French.
The fallback language is English if the current locale is not supported.
New locale language can be added by creating file message_NN.properties
in CovReport2/msg folder for the NN locale.
Feel free to provide us with these files to be included in future distributions.
Moreover, the text in the generated PDF report can be customized by modifying the message file.
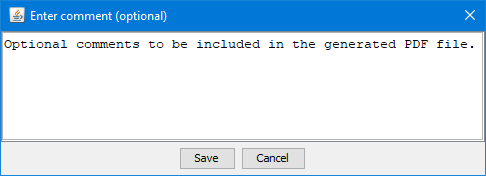
Comments
The generated PDF report could be amended with comments by clicking the button in the toolbar.
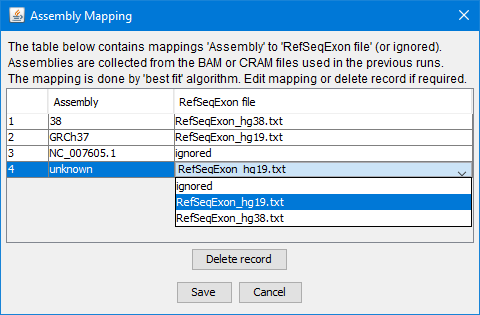
Assembly mapping to RefSeqExon file
The internal genomic interval dataset is generated by merging data from input gene/transcript list
and RefSeqExon file which serves as a dictionary.
The CovReport2 deployment includes RefSeqExon files for the genome versions hg19 and hg38:
RefSeqExon_hg19.txt
RefSeqExon_hg38.txt
The choice of RefSeqExon file is based on the assembly property in the input BAM/CRAM file header.
In certain BAM/CRAM files the assembly list is empty or contains more than one assembly name.
The application makes best efforts to find the appropriate RefSeqExon file version to use,
however certain cases may require manual matching assembly to the correct RefSeqExon file.
Click the button in the toolbar to edit assembly mapping:
A new RefSeqExon file can be added or replaced if required. It should be placed in the RefSeqExons
folder in CovReport2 deployment location.
It will be listed in the drop down option and available for manual assembly mapping.
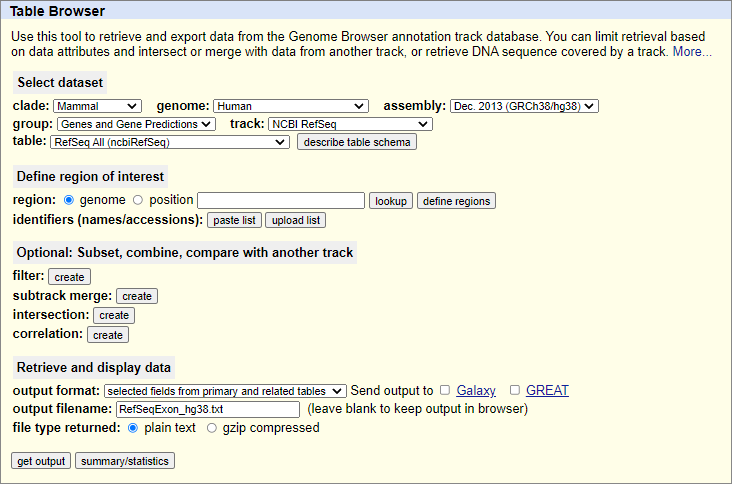
The RefSeqExon files included in the deployment were obtained from
UCSC Genome Browser
using the following input arguments (example for hg38):
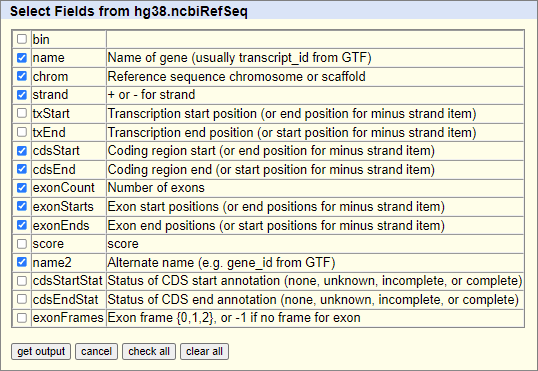
The required fields are:
Work files
For each generated report, the internal dataset can be saved for investigation or/and reuse:
bed+coverage_20x.txt - filename example for specified 20x coverage
log_success.txt or log_error.txt
The work files location is work-files/timestamp_filename-bam|cram folder.
Work files are not created by default.
Turn this feature on in Settings, see details in Configuration section.
Insertions & Deletions
The CovReport2 also includes a new experimental feature to retrieve indel data.
Information about insertions and deletions (I and D in a CIGAR string) is recorded when the BAM/CRAM file is scanned to obtain the coverage data.
Indels with read count and length above the user specified thresholds (see Configuration section) are shown.
For each genomic interval, a list of indels are specified in the following format:
COUNTx[LETTER{I,D} start length] where I and D means insertion and deletion respectively.
The below is an example of this experimental feature:
References
The following actions are available in the CovReport2 application:
Browse for input files.
Add comments to the PDF report file. This icon is disabled if no input files or patient name is provided
(see details in Comments section)
Reset application. The input files and Patient name will be cleared,
while all configuration data will remain intact.
Manually update assembly mapping
(see details in Assembly mapping section)
Open Settings popup window
(see details in Configuration section)
Open About CovReport popup window with version, build and copyright.
Updates CovReport2.config file with current configuration and closes the application.
Generates PDF report file.
This button is hidden until all required input data are provided.
Open source dependencies
The application is using open source project libraries (JARs) which are embedded into the main executable JAR:
• Htsjdk • Apache PdfBox • Apache CLI • JarClassLoader
Troubleshooting Application does not start.
• Verify that Java Runtime (JRE) is installed. Check Java version. The supported version is 8.
• Use runDebug.cmd on Windows to check the startup exception displayed in command window.
Generated PDF report file is not opened automatically.
• Verify that PDF Acrobat reader is installed.
• In rare cases the default mechanism to auto open the generated PDF file is not working.
This was observed on Mac and Linux.
Try to add the following line into the CovReport2.config file:
pdf_auto_open=N where N is a number from 0 to 4 (0 is a default).
Terms and Conditions of Use
Please find Terms and Conditions of Use document here.
You must accept and follow these Terms and Conditions of Use when using this website
and the CovReport2 application.




 in the toolbar to open Settings popup
(screenshot below for the default values):
in the toolbar to open Settings popup
(screenshot below for the default values):
 in the toolbar.
in the toolbar.
 in the toolbar to edit assembly mapping:
in the toolbar to edit assembly mapping:
 Browse for input files.
Browse for input files.
 Reset application. The input files and Patient name will be cleared,
while all configuration data will remain intact.
Reset application. The input files and Patient name will be cleared,
while all configuration data will remain intact.
 Open About CovReport popup window with version, build and copyright.
Open About CovReport popup window with version, build and copyright.
 Updates
Updates  Generates PDF report file.
This button is hidden until all required input data are provided.
Generates PDF report file.
This button is hidden until all required input data are provided.
 Application does not start.
Application does not start.